INFRECUENTES DE LA HIPOFISIS Y DE LA REGION PARASELAR
CARCINOMAS HIPOFISARIOS
A diferencia de los adenomas de hipófisis, los carcinomas de hipófisis (tumores malignos) son extremadamente raros (menos del 0.2 % de los tumores de hipófisis).
Son definidos como aquellos tumores de origen adenohipofisario que producen metástasis intracraneana o extracraneana.
En la mayoría de los casos, los carcinomas hipofisarios producen hormonas, especialmente prolactina y ACTH. Los síntomas son parecidos a los de un adenoma de hipófisis, pero también pueden incluir síntomas secundarios al crecimiento del tumor en la zona de la metástasis (por ej. convulsiones si la metástasis es a nivel cerebral).
A diferencia de otras formas de cáncer humano, las características morfológicas genéricas de malignidad; como pleomorfismo celular y nuclear, aumento de la actividad mitótica, necrosis, hemorragia, y la invasión, no son suficientes para constituir un diagnóstico de carcinoma pituitario.Los carcinomas muestran grados variables de atipia nuclear y pleomorfismo celular, pero también muestran tasas mitóticas e índices de proliferación celular significativamente más altos que los adenomas. El diagnóstico debe basarse en la presencia de al menos una metástasis intra o extracraneana. En este sentido, la diseminación metastásica parece ocurrir más a menudo dentro del sistema nervioso central a través del líquido cefalorraquídeo. Aunque una variedad de sitios metastásicos extracraneanos se han descripto (hígado, pulmones, ganglios linfáticos, corazón, huesos y riñones). El modo de difusión extracraneal se presume que es tanto hematógena y linfática.
La presentación clínica es variable. En algunos pacientes, el curso clínico inicial es indistinguible del de un adenoma pituitario típico, excepto que es seguido por las recurrencias prolongadas y la metástasis con el paso del tiempo. En otros, sin embargo, la biología del tumor maligno es evidente desde el inicio, a partir de la agresión local y progresando rápidamente a la diseminación metastásica.
El tratamiento abarca la cirugía, la radioterapia, la farmacoterapia como en los adenomas hipofisarios; pero es muy importante el rol de la quimioterapia para mejorar la supervivencia. De acuerdo con la literatura sólo el 20% sobreviven por más de 8 años.
METÁSTASIS HIPOFISARIAS
Hasta el 4% de los tumores hipofisarios son metástasis procedentes de neoplasias en otras localizaciones. Las neoplasias que con mayor frecuencia dan lugar a metástasis hipofisarias son las de mama, pulmón, tumores gastrointestinales, próstata y melanoma. Los síntomas típicos de presentación son cefalea, alteraciones visuales, diplopía y especialmente diabetes insípida. La presencia de esta última es un criterio de sospecha importante, ya que aparece hasta en el 70% de los pacientes con metástasis hipofisarias en el momento del debut clínico, mientras que aparece en menos del 1% de pacientes con adenomas hipofisarios. Debido a que la hipófisis posterior tiene un suministro directo de sangre arterial, en contraposición a la circulación portal del lóbulo anterior, más de 70% de las metástasis hipofisarias comprometen al lóbulo posterior de la hipófisis. El déficit de hormonas de la hipófisis anterior es mucho menos frecuente. La resonancia magnética puede ser útil para detectar metástasis concomitantes en el parénquima cerebral. El tratamiento es la radioterapia, sola o asociada a cirugía.
TUMOR DE CÉLULAS GRANULARES DE LA NEUROHIPÓFISIS
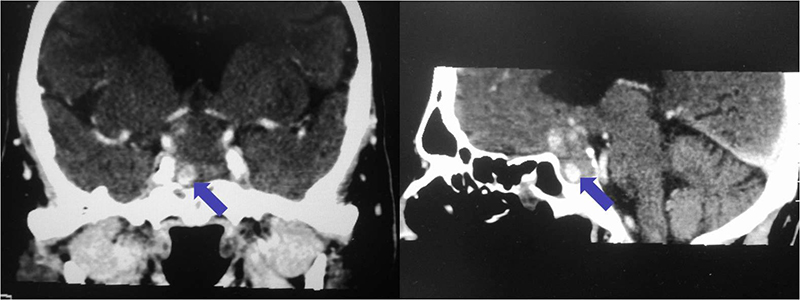
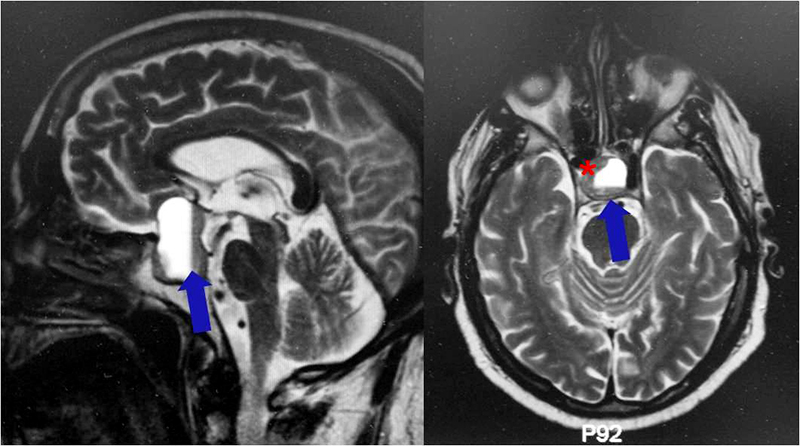
Es un tumor benigno muy infrecuente, supraselar y/o intraselar, que se origina en la neurohipófisis o en el infundíbulo. Se presenta habitualmente en la edad adulta, con mayor incidencia en mujeres. No existen síntomas o signos específicos que permitan distinguirlo de otras lesiones supraselares, aunque la diabetes insípida es relativamente infrecuente. El diagnostico final es generalmente a través de la anatomía patológica donde se constatan células con citoplasma granular eosinófilo, debido a la presencia de abundantes lisosomas. Estos tumores son immunonegativos para todas las hormonas de la hipófisis anterior, de variable positividad para la proteína S-100, y por lo general negativos para la proteína gliofibrilar ácida (GFAP). La resonancia magnética suele mostrar una lesión supraselar bien circunscrita, con captación homogénea o heterogénea de contraste. La cirugía es el tratamiento de elección.
PITUICITOMA
Se define como un tumor glial de bajo grado, muy infrecuente, compuesto por células fusiformes, sólido y bien delimitado que aparece en población adulta y que se origina en la neurohipófisis o en el infundíbulo (anteriormente denominados astrocitomas de la hipófisis posterior o infundibulomas).
Aparece en la edad adulta, y es más frecuente en hombres, con un pico de incidencia entre los 40 y 60 años de edad. Su localización puede ser intraselar, supraselar o ambas. La clínica de estos tumores comprende la alteración del campo visual, cefalea y alteración en la función hormonal hipofisaria. Las pruebas de neuroimagen muestran una lesión homogénea, bien circunscrita, con captación homogénea del contraste.
Morfológicamente, los tumores se componen de células piloides alargadas dispuestas en fascículos en un patrón que se asemeja a un astrocitoma pilocítico. A diferencia de los astrocitomas pilocíticos, sin embargo, la mayoría de pituicitomas carecen de fibras de Rosenthal y de cuerpos granulares eosinofílos. La actividad mitótica y el índice Ki67 son generalmente bajos. Por inmunohistoquímica son típicamente inmunorreactivos para vimentina, proteína S100y proteína gliofibrilar ácida (GFAP).
En los casos sintomáticos el tratamiento quirúrgico con exéresis completa ha sido el de elección, existiendo una tasa de recurrencia cercana al 20%.
ONCOCITOMA FUSOCELULAR HIPOFISARIO
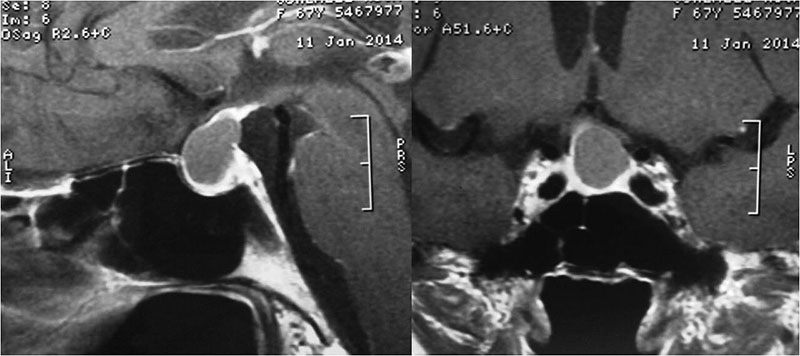
Es un tumor infrecuente (0.4%), benigno, no endocrino, originado en la adenohipófisis. Su origen es incierto, aunque se ha propuesto que deriva de las células fusiformes de la hipófisis anterior ricas en mitocondrias. Por inmunohistoquímica se obtiene positividad para proteína S-100, vimentina, galectina-3, EMA y TTF1. Aparece en la edad adulta y por igual en ambos sexos. Las características clínicas y de neuroimagen son idénticas a las de un adenoma no funcionante de hipófisis. El tratamiento de elección es la resección quirúrgica.
CORISTOMA
Son lesiones compuestas por neuronas maduras ganglionares que se encuentran adyacentes a un adenoma hipofisario, pero con un límite mal definido entre ambos. Estas neuronas contienen abundante citoplasma y gránulos de Nissl y pueden inmunomarcar para sinaptofisina, enolasa neuronal especifica y neurofilamentos. El cuadro típico histológico consiste en neuronas maduras y elementos gliales interpuestos en el fondo de un adenoma hipofisario.
Los coristomas liberan hormonas hipotalámicas, pese a no tener continuidad anatómica con el hipotálamo, lo que podría inducir la transformación de las células adenohipofisarias hacia un adenoma. Entre los casos reportados hasta la fecha, la GHRH es el producto más común, lo que resulta en un adenoma hipofisario productor de GH y una acromegalia clínica. También se han descripto coristomas productores de CRH, con la aparición un adenoma corticotropo y subsecuente enfermedad de Cushing. El tratamiento es quirúrgico y el diagnostico es retrospectivo en base a la anatomía patológica.
TUMORES INFRECUENTES PARASELARES
Existen una gran variedad de tumores que se originan en la región vecina a la glándula hipófisis, denominada región paraselar. Estos tumores son muy infrecuentes y de difícil diagnostico y tratamiento, pudiendo ser tumores de comportamiento benigno o maligno. Debido a su baja frecuencia solamente se los mencionara en la tabla a continuación.