INTRODUCCIÓN
El adenoma de hipófisis que produce y secreta hormona de crecimiento (GH) produce un cuadro clínico en el adulto denominado acromegalia. Cuando, mucho menos frecuente, el adenoma ocurre en niños o adolescentes; antes de que se detenga el crecimiento de los huesos largos, al cuadro clínico se lo denomina gigantismo y el paciente alcanza una estatura importante.
Normalmente, la GH es la responsable del crecimiento y desarrollo del cuerpo humano, especialmente durante la infancia y la adolescencia. Adicionalmente, la GH tiene funciones importantes durante la vida adulta, influyendo en el metabolismo de la glucosa y lípidos y en el desarrollo de la masa muscular y ósea. Si bien la GH tiene acciones directas sobre varios tejidos en el organismo, su principal acción la ejerce sobre el hígado. Allí, estimula la producción y secreción de una hormona denominada IGF-1 (factor de crecimiento similar a la insulina tipo 1), también conocida como Somatomedina C; la cual es la responsable final de las acciones de la GH.
En la acromegalia y el gigantismo se encuentran elevados los niveles de GH y de IGF-1, siendo este último el principal efector del cuadro clínico.
Al tipo de adenoma de hipófisis que secreta GH se lo conoce como somatotropinoma (a veces puede co-secretar PRL y se lo denomina adenoma mixto) y representa alrededor del 20% de todos los adenomas de hipófisis en la práctica médica.
Tanto la acromegalia como el gigantismo son muy infrecuentes, con una incidencia de 3 casos nuevos por millón de habitantes por año, lo que lleva a una prevalencia de 40 a 60 personas afectadas por millón de habitantes.
En raras ocasiones (menos del 2%), la acromegalia o el gigantismo pueden ser causados por un tumor localizado en otras áreas del cuerpo fuera de la glándula hipófisis (acromegalia o gigantismo extrahipofisario).
PRESENTACIÓN CLÍNICA
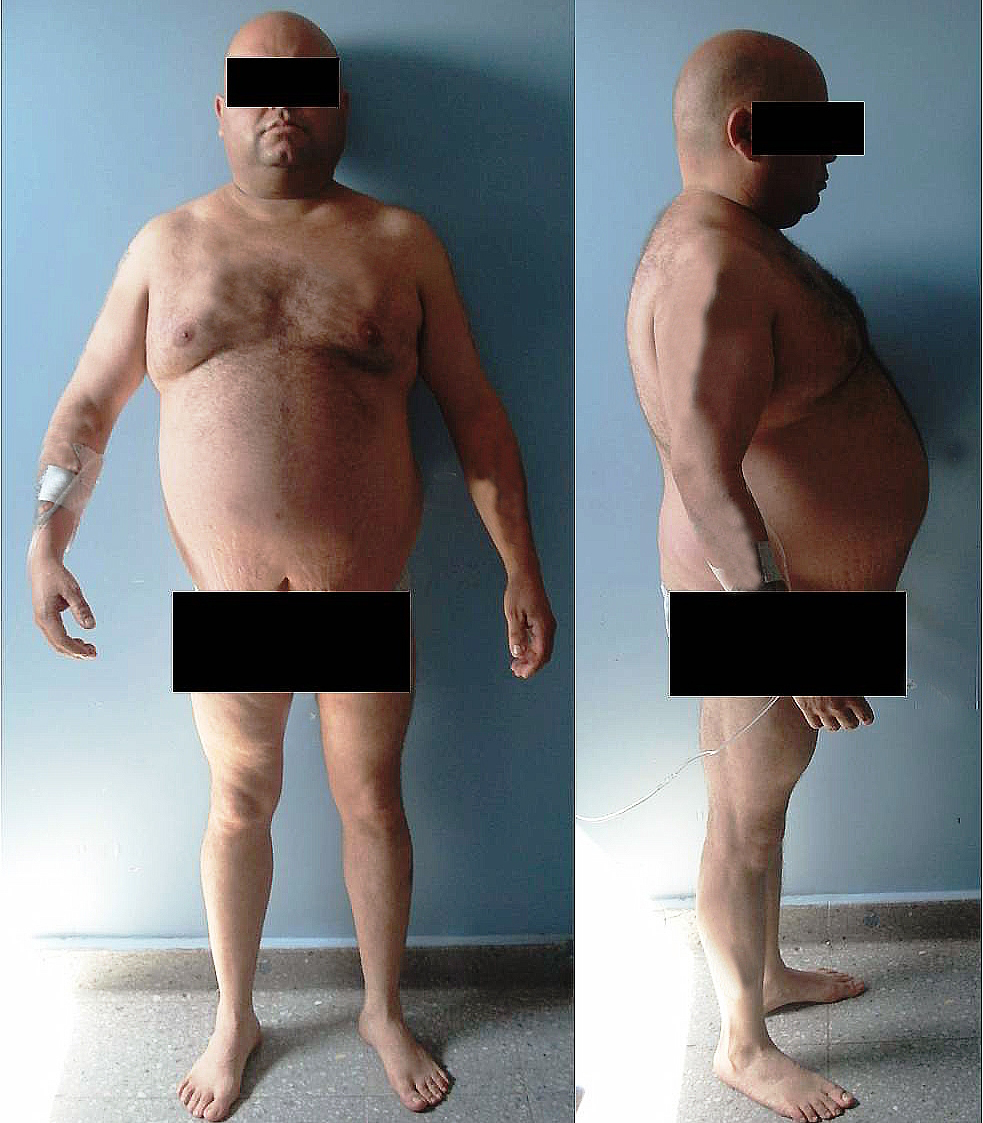
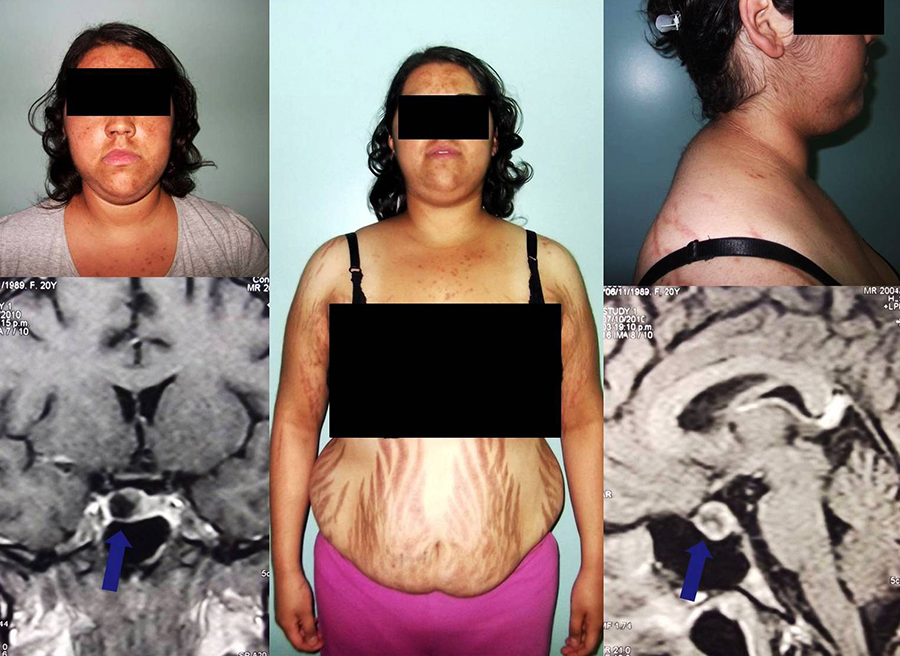
La acromegalia ocurre exclusivamente en la edad adulta, habitualmente entre los 30 y 50 años, y afecta por igual a ambos sexos. El cuadro clínico es de lenta evolución y el diagnóstico se realiza muchas veces luego de varios años desde el comienzo de los síntomas (entre 5 y 10 años). Al ser estos cambios corporales muy paulatinos; los propios pacientes, sus familiares, amigos e incluso muchos médicos piensan que estas variaciones físicas son sólo cambios naturales del envejecimiento. Es por eso que la mayoría de los pacientes pueden consultar a diferentes especialistas sin que se arribe al diagnóstico correcto. Cuando esto sucede, más del 75% de los pacientes son portadores de un macroadenoma.En los adultos ya se ha producido el cierre de los cartílagos de crecimiento de los huesos largos (normalmente a los 18 años de edad) y, por lo tanto, no existe aumento de la talla en un paciente con acromegalia.
En el gigantismo, el cuadro clínico es similar al de la acromegalia pero con la diferencia de que además existe un crecimiento excesivo de los huesos largos; lo que lleva a que el niño o el adolescente tenga una altura excesivamente mayor que la de sus pares. Esto se produce porque la GH actúa sobre los cartílagos de crecimiento ubicados en los extremos de los huesos largos (epífisis) estimulando su crecimiento excesivo. Así, el diagnóstico es más precoz que en la acromegalia debido a una altura llamativamente anormal para la edad, lo que motiva una consulta médica más temprana.
Tanto en la acromegalia como en el gigantismo el cuadro clínico está compuesto por:
Cambios fisonómicos y síntomas
-
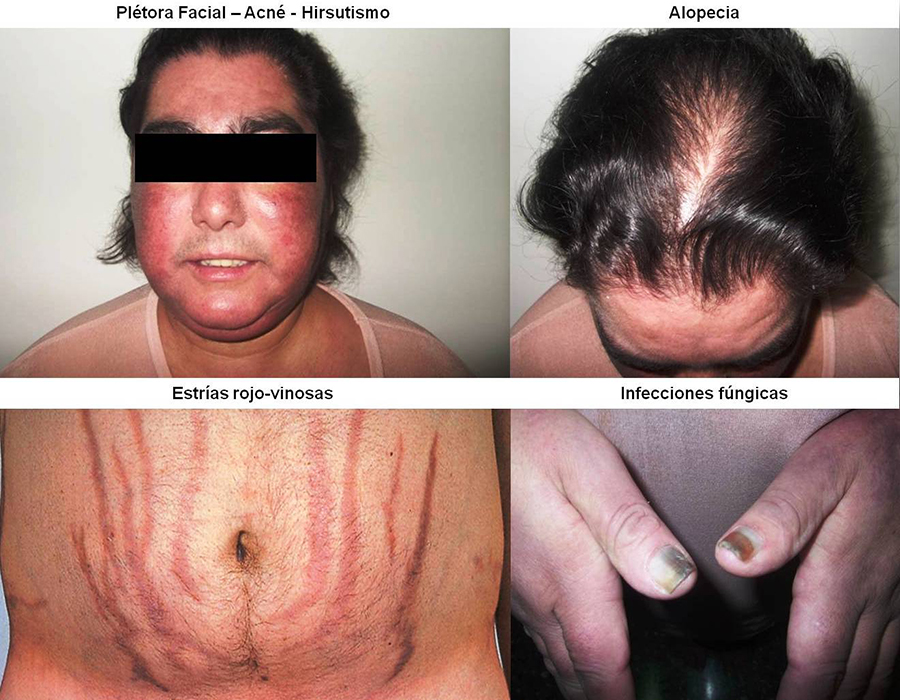
Rasgos faciales acentuados por crecimiento de los huesos faciales.
-
Aumento del tamaño del labio inferior y de la nariz.
-
Separación de las piezas dentarias (diástasis) y agrandamiento de la mandíbula (prognatismo).
-
Agrandamiento de la lengua (macroglosia).
-
Voz más grave.
-
Engrosamiento y edema de los párpados.
-
Crecimiento exagerado de las manos, pies y dedos.
-
Piel grasosa y aumento del tamaño de los poros.
-
Hiperpigmentación o acantosis nigricans.
-
Acrocordomas.
-
Sudoración excesiva (hiperhidrosis).
-
Sobrepeso u obesidad.
-
Neuropatías periféricas (síndromes del túnel carpiano u otras).
-
Artropatía: se manifiesta por dolor, inflamación y reducción de la movilidad de múltiples articulaciones.
-
Aumento excesivo de la talla corporal para la edad (únicamente en el gigantismo).
-
Retraso en la pubertad (gigantismo).
Cambios metabólicos y/o funcionales
-
Hipertensión arterial.
-
Insuficiencia cardíaca.
-
Arritmias cardíacas.
-
Aumento del tamaño de los órganos abdominales (visceromegalia).
-
Debilidad muscular y cansancio.
-
Apnea del sueño: pausas prolongadas en la respiración durante la fase de sueño.
-
Intolerancia a la glucosa o diabetes.
-
Dislipemia: hipertrigliceridemia y/o hipercolesterolemia.
-
Pólipos de colon.
-
Aumento del riesgo de desarrollar tumores en colon, próstata o glándula tiroides.
Clínica relacionada con la compresión por el adenoma
-
Dolor de cabeza (cefalea).
-
Alteración de la visión.
-
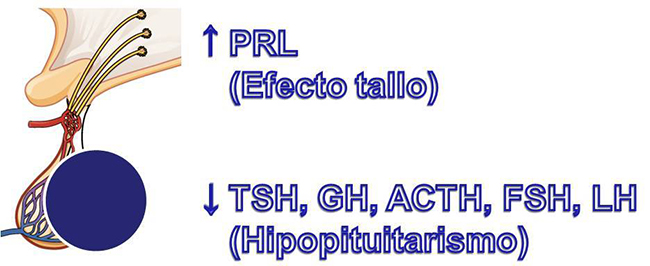
Aumento de la prolactina por "efecto tallo".
-
Disminución de una o varias hormonas producidas en la glándula hipófisis (Hipopituitarismo).

DIAGNÓSTICO
Laboratorio Hormonal
Ante la sospecha clínica de acromegalia o de gigantismo debe realizarse un examen hormonal específico en sangre para confirmar el diagnóstico.
La GH en exceso estimula al hígado para que sintetice IGF-1, el cual se encuentra elevado en la acromegalia o en el gigantismo y es la verdadera responsable del cuadro clínico.
El dosaje aislado de la GH puede llevar a imprecisiones en el diagnostico, ya que sus niveles oscilan mucho a lo largo del día, pudiendo estar normales o aumentados en el momento del dosaje. Por eso, lo correcto es medir IGF-1 ya que este es el marcador hormonal más preciso para arribar al diagnóstico correcto. Si los niveles de IGF-1 son elevados en comparación con los niveles normales para la edad y el sexo, se procederá a realizar un test dinámico denominado test de tolerancia oral a la glucosa (TTOG).
Durante esta prueba se hace ingerir al paciente una solución con 75 gramos de glucosa (azúcar) y luego se realizan extracciones de sangre para medir GH cada 30 minutos durante un lapso de 2 horas. En individuos sanos, los niveles de GH se suprimirán a menos de 1 ng/ml después de la ingestión de glucosa. Ante la presencia de acromegalia o de gigantismo los niveles de GH no se suprimirán y seguirán elevados por encima de 1 ng/ml pese a la ingesta de la glucosa.
Una vez que se realiza el diagnóstico de acromegalia o de gigantismo se debe evaluar el resto de la función hipofisaria. Por lo que se deben solicitar las siguientes hormonas: Prolactina (PRL), Tirotrofina (TSH), Triiodotironina (T3), Tiroxina (T4), Foliculoestimulante (FSH), Luteinizante (LH), Testosterona, Estrógenos, Progesterona, Adrenocorticotrofina (ACTH), Cortisol plasmático.
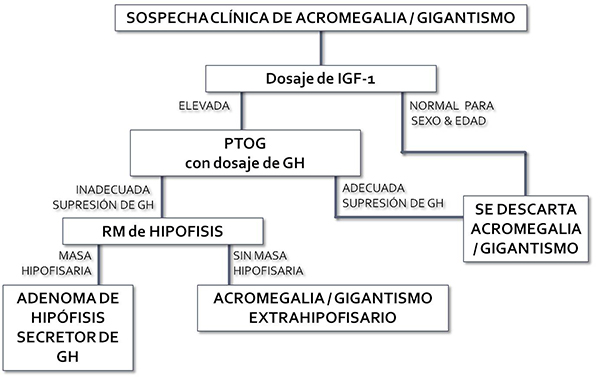
Resonancia Magnética de Cerebro Focalizada en Hipófisis con Gadolinio
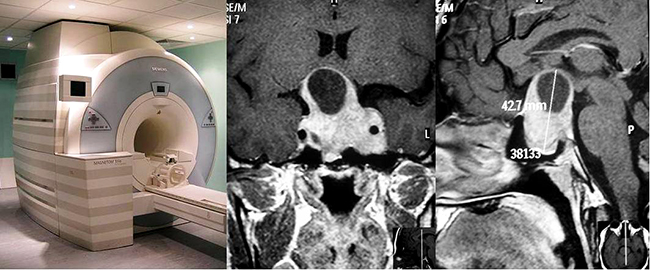
Cuando se confirma la elevación de IGF-1 y además la GH no suprime durante el test de tolerancia oral a la glucosa, el siguiente paso es realizar una resonancia magnética con foco en la glándula hipófisis. Así, podrá evaluarse si se está en presencia de un microadenoma o de un macroadenoma. Este estudio es fundamental ya que certifica el diagnóstico y además permite evaluar si el adenoma se encuentra invadiendo las estructuras que lo rodean (por ejemplo, el seno cavernoso) o bien si tiene una extensión supraselar importante. Estos factores son de vital importancia para saber el grado de respuesta a una eventual cirugía.
Muy raramente (menos del 2%), la resonancia puede no mostrar un adenoma de hipófisis. Estos casos son definidos como causas extrahipofisarias de acromegalia o de gigantismo y requieren otro tipo de estudios complementarios.
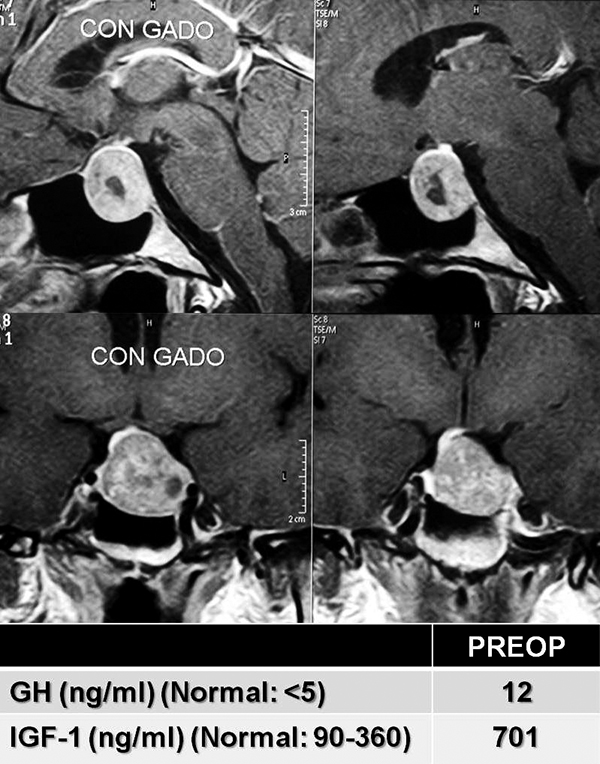
|
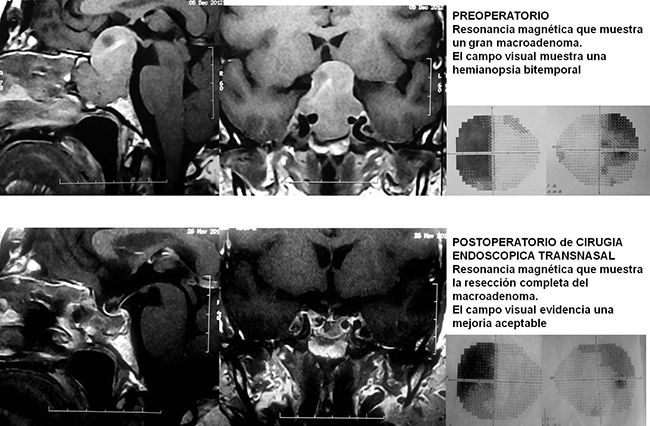
Resonancia magnética preoperatoria que muestra un macroadenoma productor de GH.
Se muestran también los valores de GH e IGF-1 elevados antes de la cirugía transnasal endoscópica.
|
Campo Visual Computarizado
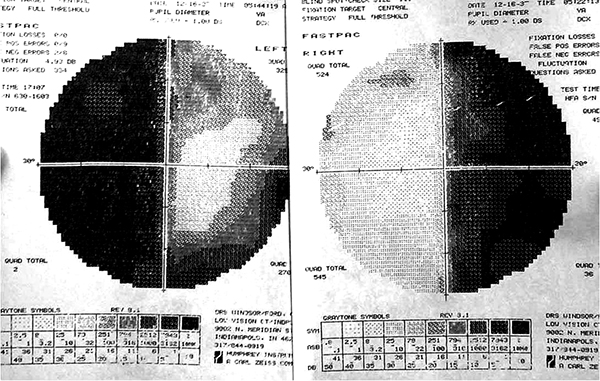
En el caso de los macroadenomas es necesario determinar si existe algún compromiso en la vía óptica (quiasma óptico, nervios ópticos, cintilla óptica). El cuadro típico de pérdida del campo visual se conoce como hemianopsia bitemporal y se produce por una compresión del quiasma óptico por el macroadenoma. La hemianopsia bitemporal se caracteriza por una falta de la visión periférica más lateral en ambos ojos y los pacientes solamente pueden ver lo que está justo frente a ellos. Pueden existir otro tipos de déficits visuales que van desde cuadrantopsias hasta la amaurosis (ceguera).
TRATAMIENTO
La acromegalia o el gigantismo requieren de un tratamiento efectivo para mejorar los síntomas del paciente, su calidad de vida y su expectativa de vida. Aquellos pacientes no tratados o incorrectamente controlados tienen una menor calidad de vida y su sobrevida se ve acortada con respecto a la población sana.
Alcanzar niveles de GH e IGF 1 normales reduce la mortalidad de los pacientes y la iguala con la población sana.
Existen varios pilares en el tratamiento: la cirugía, el tratamiento farmacológico y la radioterapia. Muchos pacientes requieren de la combinación de estas modalidades para un adecuado control de la enfermedad.
Objetivos del tratamiento
-
Normalización de los valores de GH.
-
Normalización de valores de IGF-1, según edad y sexo.
-
Estabilización o reducción del tamaño tumoral.
-
Mantenimiento o recuperación de la función hipofisaria.
-
Reducción de la morbimortalidad.
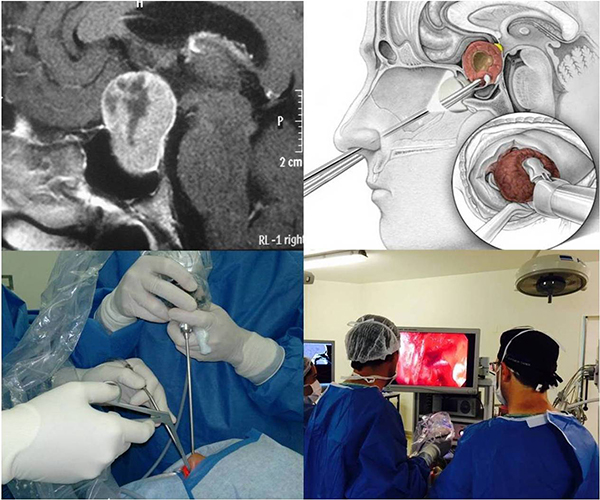
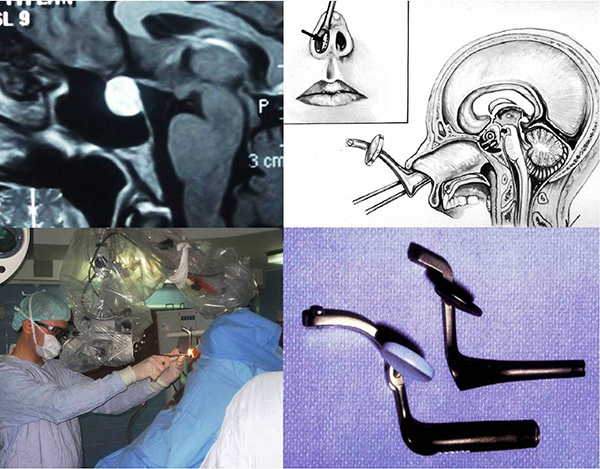
Cirugía transnasal transesfenoidal
La técnica habitual se realiza, en la inmensa mayoría de los casos (96%), a través de la nariz en lo que se denomina cirugía transnasal transesfenoidal. La misma puede ser realizada con técnicas microquirúrgicas o por endoscopia, dependiendo de cada caso.La técnica endoscópica es la de mayor difusión en la actualidad.
Menos de un 4% de los casos deben ser operados a través de una craneotomía mínimamente invasiva, es decir a través de una pequeña ventana ósea que se realiza en el cráneo. Este tipo de cirugía tiene un grado mayor de complejidad que la cirugía transnasal transesfenoidal.
La cirugía puede ser el único tratamiento necesario y lograr controlar la enfermedad, especialmente en aquellos adenomas que no invaden el seno cavernoso y/o estructuras supraselares. La cirugía tiene una tasa de éxito que depende del tamaño y grado de invasión del adenoma. Así, la cirugía puede normalizar los niveles de IGF-1 en aproximadamente el 85% de los microadenomas y en el 62% de los macroadenomas que no invaden senos cavernosos. Es por ello que sigue siendo considerada como la primera opción de tratamiento frente a un paciente con acromegalia o gigantismo. Se debe tener en cuenta que la tasa de éxito quirúrgico y el riesgo de complicaciones están relacionados directamente con la experiencia del neurocirujano.
La cirugía brinda la posibilidad de revertir muchos de los síntomas preoperatorios ocasionados por el adenoma somatotropo. Es importante saber que si el tumor puede resecarse de manera completa y el paciente reúne criterios bioquímicos de remisión, se reduce significativamente la posibilidad de recurrencia. Incluso, si la resección del adenoma es parcial y queda solo un pequeño remanente, el mismo puede controlarse más fácilmente mediante tratamiento farmacológico o por radioterapia.
En algunos pacientes puede intentarse antes de la cirugía un tratamiento con fármacos análogos de la somatostatina (tratamiento primario), con el objetivo de mejorar los síntomas antes de la cirugía e incluso, disminuir el tamaño del adenoma. Esta decisión debe ser tomada en conjunto entre el neurocirujano y el endocrinólogo en cada caso en particular.
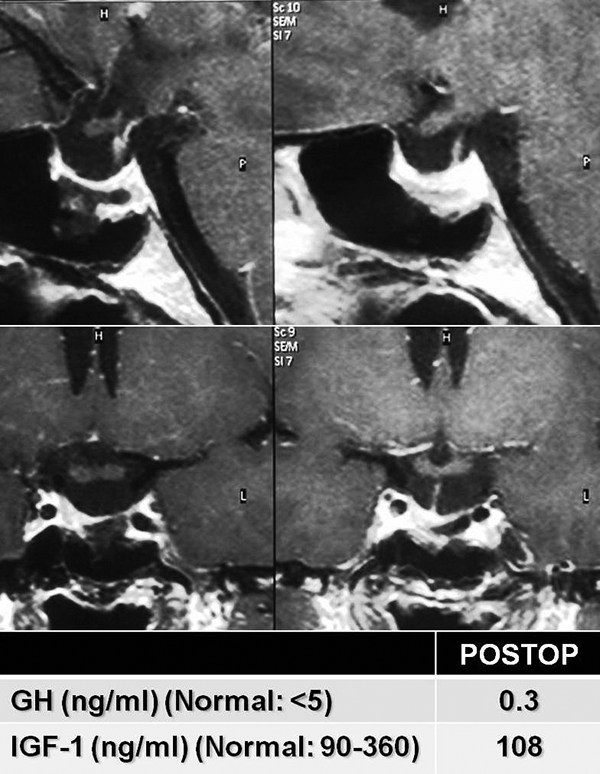
|
Resonancia magnética postoperatoria del mismo paciente que en la figura anterior, que muestra la resección completa del macroadenoma luego de una cirugía transnasal endoscópica y la identificación de la glándula hipófisis normal y el tallo hipofisario. Se muestran también los valores de GH e IGF-1 como se normalizaron luego de la cirugía. |
Tratamiento farmacológico
En líneas generales, el tratamiento farmacológico se emplea luego de que el paciente se ha sometido a una cirugía y en donde la enfermedad se encuentra aún activa por algún remanente de adenoma (tratamiento secundario).
Independientemente del fármaco empleado, es necesario saber que el tratamiento es por tiempo indefinido, muy probablemente de por vida; y que no se sugiere discontinuar el mismo debido al riesgo de recurrencia de la enfermedad.
Existen tres grupos de fármacos disponibles:
Análogos de la somatostatina
La somatostatina es una hormona que normalmente inhibe la secreción de GH. Aquellos fármacos denominados "análogos de la somatostatina” reproducen la acción inhibitoria de la somatostatina y han sido desarrollados para disminuir los niveles de GH y de IGF-1 en pacientes con acromegalia o gigantismo, actuando directamente sobre el tumor. Estos fármacos pueden también reducir el tamaño del adenoma somatotropo.
En líneas generales, los análogos de la somatostatina pueden normalizar los niveles de IGF-1 en aproximadamente el 50% de los pacientes y también pueden disminuir el tamaño tumoral hasta en un 50% de los casos.
Los análogos de la somatostatina disponibles y aprobados en la actualidad son:
-
Octreotide SC: aplicación inyectable subcutánea (3 veces por día). Dosis de 150 mcg a 1.5 mg por día.
-
Octreotide LAR: aplicación inyectable intramuscular. Dosis de 10 a 40 mg cada 28 días.
-
Lanreotide SR: aplicación inyectable intramuscular. Dosis de 30 mg cada 10-14 días.
-
Lanreotide ATG: aplicación inyectable subcutánea. Dosis de 60 a 120 mg cada 28-56 días.
Los efectos secundarios más comunes de análogos de la somatostatina incluyen náuseas, diarrea, molestias abdominales y el desarrollo de cálculos biliares. Un porcentaje de pacientes desarrolla intolerancia a la glucosa o diabetes.
Agonistas dopaminérgicos
Los agonistas de la dopamina pueden disminuir la secreción de GH y IGF-1 actuando directamente sobre las células tumorales. Tienen la ventaja de administrarse por vía oral y ser bien tolerados; sin embargo, son menos efectivos que los análogos de la somatostatina para el control de la enfermedad si se utilizan como única droga. Así, pueden normalizar los niveles de IGF-1 y GH en el 15% de los pacientes.
Por tal motivo, pueden combinarse con algún análogo de la somatostatina para aumentar la efectividad del tratamiento.
Los fármacos aprobados mundialmente son:
-
Cabergolina. Dosis de 1 a 4 mg semanalmente.
-
Bromocriptina. Dosis de 2.5 mg a 7.5 mg por día
Antagonista del receptor de GH
El Pegvisomant es un fármaco que bloquea los receptores de GH a nivel hepático y por lo tanto disminuye los niveles de IGF-1 en sangre y también actúa en el resto de los tejidos bloqueando la acción de la GH. Utilizado como única droga puede normalizar los niveles de IGF-1 en aproximadamente el 60% de los pacientes. Además, también puede utilizarse solo o en combinación con análogos de la somatostatina o con agonistas dopaminérgicos.
-
Pegvisomant: aplicación inyectable subcutánea. Dosis de 10 a 30 mg por día.
Fármacos en vía de aprobación
Pasireotide LAR: Es un nuevo análogo de la somatostatina, con mayor control bioquímico que los análogos de lasomatostatina convencionales (por ej. Octreotide LAR). Los efectos adversos son similares al resto de los análogos de la somatostatina con mayor riesgo de hiperglucemia o diabetes.
Fármacos aun no aprobados pero que se encuentran en fase de investigación
-
Octreotide oral.
-
Octreotide subcutáneo de larga acción.
-
Implantes de Octreotide de larga acción.
Terapia de reemplazo hormonal
Como parte del tratamiento integral se debe realizar el correcto reemplazo del resto de las hormonas hipofisarias que se hayan dañado por efecto directo del tumor o como consecuencia de la cirugía y/o radioterapia, ya que ello impacto en la calidad de vida y en la mortalidad a largo plazo. Para ello es el endocrinólogo el que debe controlar periódicamente el nivel de las hormonas hipofisarias.
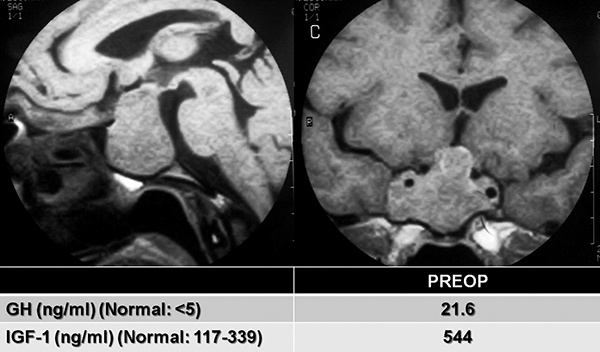
|
Resonancia magnética preoperatoria que muestra un gran macroadenoma productor de GH que invade ambos senos cavernosos y engloba a ambas arterias carótidas (Knosp IV). Se muestran también los valores de GH e IGF-1 elevados antes de la cirugía. |

|
Resonancia magnética postoperatoria del mismo paciente que la figura anterior. Luego de una cirugía transnasal endoscópica se logró una resección medial del macroadenoma, con persistencia previsible de restos de adenoma en ambos senos cavernosos (asteriscos). Se muestran también los valores de GH e IGF-1 que persisten elevados pese a la cirugía, aunque con un descenso marcado con respecto a los valores preoperatorios. Este paciente requirió de un tratamiento farmacológico con Octreotide LAR 40 mg mensuales y Cabergolina 3 mg semanales para poder controlar su enfermedad.
|
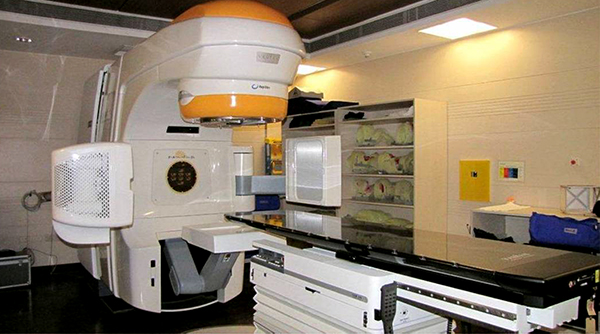
Radioterapia
En el caso de los adenomas somatotropos que producen acromegalia o gigantismo, la utilización de la radioterapia queda reservada exclusivamente para aquellos casos en donde la enfermedad sigue activa a causa de algún remanente de adenoma, el cual no pudo ser controlado por la cirugía ni por un adecuado tratamiento farmacológico. La radioterapia hipofisaria nunca es la primera línea de tratamiento en la acromegalia o en el gigantismo, y debe ser criteriosamente indicada a través de un consenso entre el endocrinólogo y el neurocirujano. Además, hay que tener en cuenta que el resultado de la radioterapia puede observarse luego de varios años de realizada la misma, por lo que el paciente durante ese lapso deberá continuar con el tratamiento farmacológico. Las modalidades de radioterapia en la actualidad son: la radiocirugía estereotáctica o bien la radioterapia estereotáctica fraccionada.